共定位分析Qtls-Gwas
共定位分析Qtls-Gwas.Rmd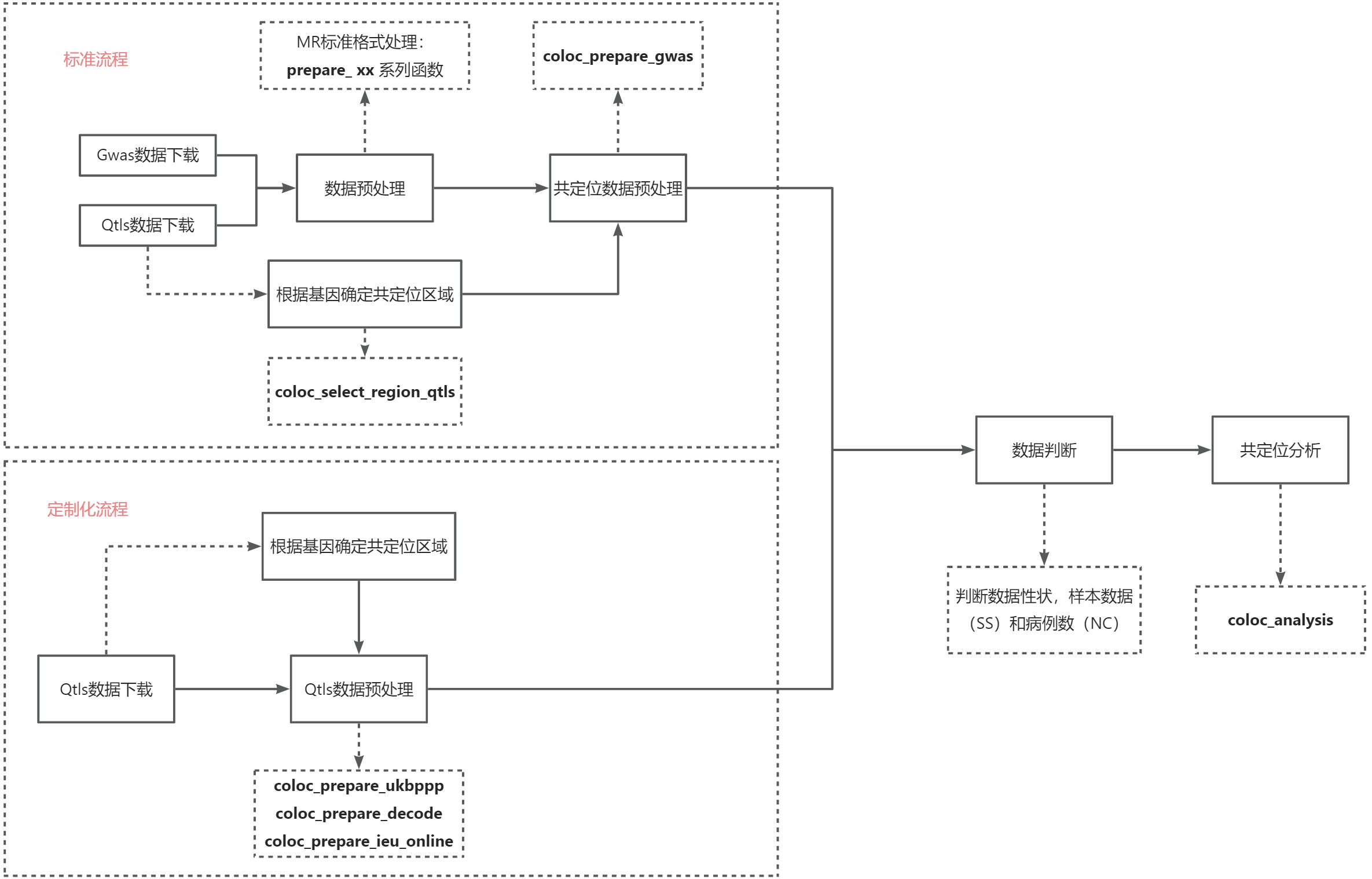
1. 标准化流程
1.1. 前言
Qtls-gwas共定位分析共定位主要分为以下几步,以GCKR 和
T2D_WIDE 数据示例:
step1:GWAS数据预处理,转换成标准MR格式
step2:Qtls数据预处理,转换成标准MR格式
step3:根据基因确定共定位区域
step4:Gwas、Qtls数据共定位预处理,筛选选定的共定位区域数据
-
step5:共定位分析,提前判断数据类型,并提供相关数据进行分析
quant(数量性状)数据,只需要提供样本数据(SS)
cc(二分类性状)数据,需要提供样本数据(SS)和病例数(NC)
1.2. 数据预处理
在执行本地数据进行分析的时候,均需要按照标准数据准备流程进行预处理,参考:MR标准格式。
1.2.1. GWAS数据下载及预处理
下载数据👉:finngen_R9_T2D_WIDE.gz
prepare_finngen(
file_path = "./finngen_R9_T2D_WIDE.gz",
out_path = "./"
)由上可得到:finngen_R9_T2D_WIDE.txt 标准MR格式文件
1.2.2. Qtls下载
在此,使用decode-2021来源数据,需要自己填写申请,官网将下载链接发送到邮件:
Ferkingstad, E. et al. Large-scale integration of the plasma proteome with genetics and disease. Summary data (list). 👈点此可申请下载
以GCKR基因为例,在decode-2021下载获得5223_59_GCKR_GCKR.txt.gz数据
prepare_decode(
file_path= "./5223_59_GCKR_GCKR.txt.gz",
snp_annotation_path = "./assocvariants.annotated.index",
out_path = "./"
)由上可得到:5223_59_GCKR_GCKR.txt 标准MR格式文件
备注:assocvariants.annotated.index
由我们构建的索引文件,方便处理快速的处理decode-2021数据。(联系客服获取)
1.2.3. bfile数据下载
bfile数据下载: http://fileserve.mrcieu.ac.uk/ld/1kg.v3.tgz
1.3. 共定位区域选择
以GCKR基因为例,根据基因查询选择筛选数据范围,Ensembl_GRCh38已内置到R包中。
# pQTLs用cis区域的QTLs数据,即编码蛋白基因区域的上下游1m范围之内
# 获取GCKR的cis-pQTLs
temp <- Ensembl_GRCh38[Ensembl_GRCh38$gene_name=="GCKR",]
coloc_select_region_qtls(
gene_chr = temp$seqnames,
gene_start = temp$start,
gene_end = temp$end,
up_num = 1e6,
down_num = 1e6,
out_path = "./",
out_prefix = "qtls_GCKR"
)
# ✔ 用于coloc分析的染色体区域文件的输出路径为: ./qtls_GCKR_coloc_region.txt
# $chrompos
# [1] "2:26496839-28523684"
#
# $start
# [1] 26496839
#
# $end
# [1] 28523684
#
# $chr
# [1] "2"由上可得到:qtls_GCKR_coloc_region.txt
当前基因的共定位筛选范围
1.4. 共定位数据预处理
根据区域筛选(标准mr文件)数据,并输出共定位标准文件。
即:标准mr文件 —> 筛选区域 —> 输出标准共定位文件
# 筛选范围与qtls一样的范围数据
region <- readLines("./qtls_GCKR_coloc_region.txt")
chrompos <- region
coloc_prepare_gwas(file_path = "./finngen_R9_T2D_WIDE.txt",
chrompos = chrompos,
out_path = "./",
out_prefix = "gwas"
)
# ✔ 用于coloc分析的文件输出到: ./gwas_coloc_2-26496839-28523684.txt
# chrompos nrows file_path
# 1 2:26496839-28523684 10805 ./gwas_coloc_2-26496839-28523684.txt
coloc_prepare_gwas(file_path = "./5223_59_GCKR_GCKR.txt",
chrompos = chrompos,
out_path = "./",
out_prefix = "qtls_GCKR"
)
# ✔ 用于coloc分析的文件输出到: ./qtls_GCKR_coloc_2-26496839-28523684.txt
# chrompos nrows file_path
# 1 2:26496839-28523684 12124 ./qtls_GCKR_coloc_2-26496839-28523684.txt由上可得到,根据区域筛选的数据文件:
gwas_coloc_2-26496839-28523684.txtqtls_GCKR_coloc_2-26496839-28523684.txt
1.5. 共定位分析
根据共定位预处理输出文件进行分析,根据样本类型,提供原始数据样本数据(SS)或病例数(NC)。相关数据内容,可以查询数据来源。
- 5223_59_GCKR_GCKR 是quant(数量性状)数据,只需要提供样本数据(SS)。
# 读取预处理的decode数据,方便后续查询样本数和病例数
GCKR <- data.table::fread("./qtls_GCKR_coloc_2-25496839-29523684.txt")
max(GCKR$samplesize)
# gwas1:
# SS1 数据字段:samplesize 字段(数字偏差不大,即可使用)- finngen_R8_T2D_WIDE 是cc(二分类性状)数据,需要提供样本数据(SS)和病例数(NC)。
# 读取finngen数据来源,方便后续查询样本数和病例数
finngen_list <-data.table::fread("./R9_manifest.tsv",data.table = T)
finngen_list <- finngen_list[grepl("finngen_R9_T2D_WIDE",finngen_list$path_bucket),]
# gwas2:
# finngen_list 查询finngen_R9_T2D_WIDE,找到样本数据(SS)和病例数(NC)
# SS2 数据字段: num_controls
# NC2 数据字段: num_cases- 分析
coloc_analysis(
gwas_path1 = "./qtls_GCKR_coloc_2-26496839-28523684.txt",
type1 = "quant",
SS1 = 35377,
NC1 = 0,
gwas_path2 = "./gwas_coloc_2-26496839-28523684.txt",
type2 = "cc",
SS2 = 284971,
NC2 = 33043,
run_coloc_susie = T,
bfile = "./1kg.v3/EUR",
out_path = "./",
out_prefix = "qtls-gwas"
)
# 可不执行coloc_susie分析
# coloc_analysis(
# gwas_path1 = "./data_prepare/qtls_GCKR_coloc_2-25496839-29523684.txt",
# type1 = "quant",
# SS1 = 35377,
# NC1 = 0,
# gwas_path2 = "./data_prepare/gwas_coloc_2-27496839-27523684.txt",
# type2 = "cc",
# SS2 = 284971,
# NC2 = 33043,
# run_coloc_susie = F,
# out_path = "./result",
# out_prefix = "qtls-gwas"
# )
# ✔ 用于绘图的数据输出到: ./qtls_GCKR_coloc_2-26496839-28523684_plot.txt,./gwas_coloc_2-26496839-28523684_plot.txt
# PP.H0.abf PP.H1.abf PP.H2.abf PP.H3.abf PP.H4.abf
# 9.51e-24 6.72e-06 9.80e-21 5.93e-03 9.94e-01
# [1] "PP abf for shared variant: 99.4%"
# ✔ coloc.abf共定位分析的结果输出到: ./qtls-gwas_qtls_GCKR_gwas_coloc_abf_summary.csv,./qtls-gwas_qtls_GCKR_gwas_coloc_abf_results.csv
#✔ coloc.susie共定位分析的结果输出到: ./qtls-gwas_qtls_GCKR_gwas_coloc_susie_summary.csv,./qtls-gwas_qtls_GCKR_gwas_coloc_susie_results.csv上述示例得到以下几个文件:
qtls_GCKR_coloc_2-26496839-28523684_plot.txtgwas_coloc_2-26496839-28523684_plot.txtqtls-gwas_qtls_GCKR_gwas_coloc_abf_summary.csvqtls-gwas_qtls_GCKR_gwas_coloc_abf_results.csv
如果设置run_coloc_susie = T,成功的情况下得到:
qtls-gwas_qtls_GCKR_gwas_coloc_susie_summary.csvqtls-gwas_qtls_GCKR_gwas_coloc_susie_results.csv
1.6. 结果解读
以
_plot结尾的文件,是用于coloc_plot_locuscompare 及 coloc_plot_stack_assoc 绘图数据以
_summary结尾文件是统计结果,如果PP.H4.abf >0.95或按照自己需求适当放宽需求,那么认为此次分析有效。-
以
_results结尾文件是最终选定的snp点,在_summary有效的情况下,_results文件中SNP.PP.H4越接近1,则此位置共定位点snp。https://zhuanlan.zhihu.com/p/392589375
通常情况下,很多文献认为PPA>0.95的位点是共定位位点,也有一些文献会放松要求到0.75。
2. 定制化流程
2.1 前言
Qtls-gwas共定位分析共定位主要分为以下几步,以GCKR 和
T2D_WIDE 数据示例:
step1:GWAS数据预处理,转换成标准MR格式
step2:根据基因确定共定位区域,并预处理Qtls数据,筛选出Qtls共定位数据
step3:Gwas定位预处理,筛选选定的共定位区域数据
-
step4:共定位分析,提前判断数据类型,并提供相关数据进行分析
quant(数量性状)数据,只需要提供样本数据(SS)
cc(二分类性状)数据,需要提供样本数据(SS)和病例数(NC)
2.2 数据预处理
Gwas数据按照 1.2.1. GWAS数据下载及预处理 处理即可,Qtls数据不需要预先处理成MR格式。
2.3 共定位区域选择及Qtls数据处理
以GCKR基因为例,根据基因查询选择筛选数据范围,Ensembl_GRCh38已内置到R包中。
# pQTLs用cis区域的QTLs数据,即编码蛋白基因区域的上下游1m范围之内
# 获取GCKR的cis-pQTLs
temp <- Ensembl_GRCh38[Ensembl_GRCh38$gene_name=="GCKR",]
coloc_prepare_decode(file_path= "./5223_59_GCKR_GCKR.txt.gz",
gene_chr = temp$seqnames,
gene_start = temp$start,
gene_end = temp$end,
up_num =1e6,
down_num = 1e6,
out_path = "./data_prepare",
out_prefix = "qtls_GCKR")
# ✔ 用于coloc分析的文件输出到: ./qtls_GCKR_coloc_2-26496839-28523684.txt
# $file_path
# [1] "./qtls_GCKR_coloc_2-26496839-28523684.txt"
# $chrompos
# [1] "2:26496839-28523684"由上可得出两个文件:
qtls_GCKR_coloc_region.txt当前基因的共定位筛选范围qtls_GCKR_coloc_2-26496839-28523684.txt根据区域筛选的数据
2.4 Gwas数据预处理
根据区域筛选(标准mr文件)数据,并输出共定位gwas标准文件。
即:标准mr文件 —> 筛选区域 —> 输出标准共定位文件
# 筛选范围与qtls一样的范围数据
region <- readLines("./qtls_GCKR_coloc_region.txt")
chrompos <- region
coloc_prepare_gwas(file_path = "./finngen_R9_T2D_WIDE.txt",
chrompos = chrompos,
out_path = "./"
)
# ✔ 用于coloc分析的文件输出到: ./gwas_coloc_2-26496839-28523684.txt
# chrompos nrows file_path
# 1 2:26496839-28523684 10805 ./gwas_coloc_2-26496839-28523684.txt由上可得到,根据区域筛选的数据文件:
gwas_coloc_2-26496839-28523684.txt