共定位分析Gwas-Gwas
共定位分析Gwas-Gwas.Rmd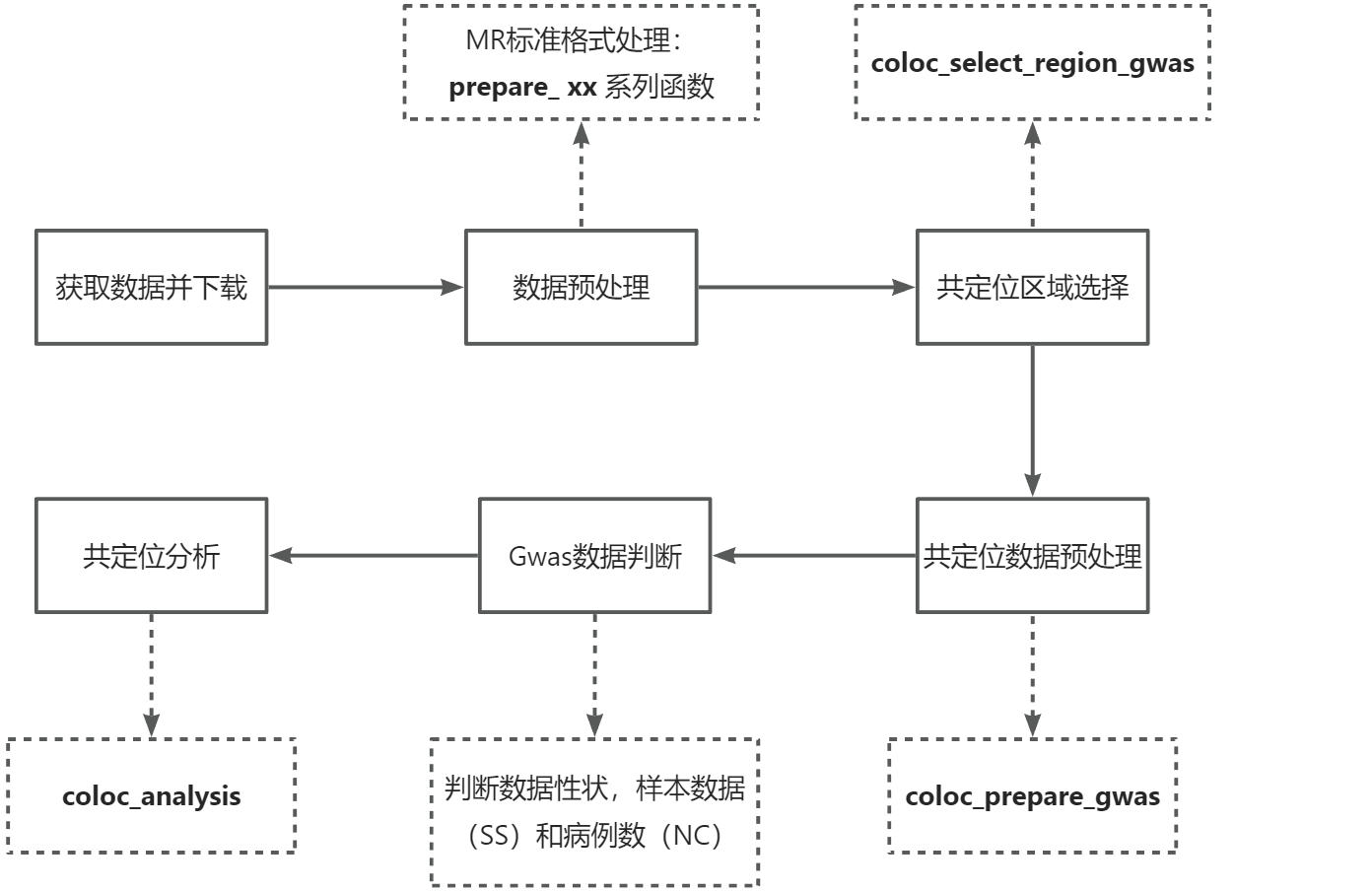
1. 前言
Gwas-gwas共定位分析主要分为四步,我们以ieu-a-300 和
ieu-a-7 数据示例:
step1:数据预处理,转换成标准MR格式
step2:共定位区域选择,选择符合P值的基因数据,显著性最高的区域
step3:共定位gwas数据预处理,筛选选定的共定位区域数据
-
step4:共定位分析,提前判断数据类型,并提供相关数据进行分析
quant(数量性状)数据,只需要提供样本数据(SS)
cc(二分类性状)数据,需要提供样本数据(SS)和病例数(NC)
2. Gwas数据下载及预处理
在执行本地数据进行分析的时候,均需要按照标准数据准备流程进行预处理,参考:MR标准格式,用以下代码完成标准准备:
2.2 数据预处理
prepare_ieu(file_path = "ieu-a-300.vcf.gz", out_path="./")
prepare_ieu(file_path = "ieu-a-7.vcf.gz",out_path="./")3. 共定位区域选择
共定位区域选择,需要筛选符合p值范围的基因数据。最终取p值显著性高的区域进行共定位分析(此处选择其中一个示例)。
selectd_data <- coloc_select_region_gwas(file_path = "./ieu-a-300.txt",
exp_p = 5e-08,
bfile = "./1kg.v3/EUR",
up_num = 150000,
down_num = 150000,
out_path = "./")
# 读取共定位区域,选择top显著性相关的snp,并指定用于共定位研究的染色体区域
selectd_data <- selectd_data$chrompos
# 更加p值排序,选择显著性最高的
selectd_data <- selectd_data[order(selectd_data$pval.exposure,decreasing = F) ,]
chrompos <- selectd_data$chrompos[1]
print(chrompos)
# [1] "1:109664880-109964880"可得出当前共定位区域为:1:109664880-109964880
4. 共定位Gwas数据预处理
根据区域筛选数据(标准mr文件),找到共同区域的数据,并输出共定位gwas标准文件。
即:标准mr文件 ---> 筛选区域 ---> 输出标准共定位文件
此处使用前文得到的共定位区域:1:109664880-109964880
# 共定位数据预处理
coloc_prepare_gwas(
file_path = "./ieu-a-7.txt",
chrompos = "1:109664880-109964880",
out_path = "./",
out_prefix = "gwas_coloc_7"
)
# ✔ 用于coloc分析的文件输出到: ./gwas_coloc_7_coloc_1-109664880-109964880.txt
# chrompos nrows file_path
# 1 1:109664880-109964880 745 ./gwas_coloc_7_coloc_1-109664880-109964880.txt
coloc_prepare_gwas(
file_path = "./ieu-a-300.txt",
chrompos = "1:109664880-109964880",
out_path = "./",
out_prefix = "gwas_coloc_300"
)
# ✔ 用于coloc分析的文件输出到: ./gwas_coloc_300_coloc_1-109664880-109964880.txt
# chrompos nrows file_path
# 1 1:109664880-109964880 428 ./gwas_coloc_300_coloc_1-109664880-109964880.txt由上可得到两个相同区域内数据的文件
gwas_coloc_7_coloc_1-109664880-109964880.txtgwas_coloc_300_coloc_1-109664880-109964880.txt
5. 共定位分析
根据共定位预处理输出文件进行分析,根据样本类型,提供原始数据样本数据(SS)或病例数(NC)。相关数据内容,可以查询gwas数据来源。
例如:
-
ieu-a-300:quant(数量性状)数据,只需要提供样本数据(SS)。
-
https://gwas.mrcieu.ac.uk/datasets/ieu-a-300/
- SS1数据字段: Sample size
-
-
ieu-a-7:cc(二分类性状)数据,需要提供样本数据(SS)和病例数(NC)。
-
https://gwas.mrcieu.ac.uk/datasets/ieu-a-7/
SS2 数据字段: Sample size
NC2 数据字段: ncase
-
coloc_analysis(
gwas_path1 = "./gwas_coloc_300_coloc_1-109664880-109964880.txt",
type1 = "quant",
SS1 = 173082,
NC1 = 0,
gwas_path2 = "./gwas_coloc_7_coloc_1-109664880-109964880.txt",
type2 = "cc",
SS2 = 184305,
NC2 = 60801,
out_path = "./",
out_prefix = "qtls-gwas"
)
# 可不执行coloc_susie分析
# coloc_analysis(
# gwas_path1 = "./data_prepare/gwas_coloc_300_coloc_1-109772166-109872166.txt",
# type1 = "quant",
# SS1 = 173082,
# NC1 = 0,
# gwas_path2 = "./data_prepare/gwas_coloc_7_coloc_1-109772166-109872166.txt",
# type2 = "cc",
# SS2 = 184305,
# NC2 = 60801,
# run_coloc_susie = T,
# bfile = "./data/1kg.v3/EUR",
# out_path = "./result",
# out_prefix = "qtls-gwas"
# )上述示例得到以下几个文件:
gwas_coloc_300_coloc_1-109664880-109964880_plot.txtgwas_coloc_7_coloc_1-109664880-109964880_plot.txtqtls-gwas_gwas_gwas_coloc_abf_results.csvqtls-gwas_gwas_gwas_coloc_abf_summary.csv
6. 结果解读
以
_plot结尾的文件,是用于coloc_plot_locuscompare 及 coloc_plot_stack_assoc 绘图数据以
_summary结尾文件是统计结果,如果PP.H4.abf >0.95或按照自己需求适当放宽需求,那么认为此次分析有效。-
以
_results结尾文件是最终选定的snp点,在_summary有效的情况下,_results文件中SNP.PP.H4越接近1,则此位置共定位点snp。https://zhuanlan.zhihu.com/p/392589375
通常情况下,很多文献认为PPA>0.95的位点是共定位位点,也有一些文献会放松要求到0.75。