SMR基础
SMR基础.Rmd1. SMR程序
官网👉SMR 下载即可,或根据当前操作系统点击下方链接下载
- Linux:smr-1.3.1-linux-x86_64.zip
- Mac:smr_Mac_v1.03.zip
- Windows:smr-1.3.1-win-x86_64.zip
注意:下载之后,解压保存到固定的文件目录,后续函数使用指定其可执行文件全路径即可
2. SMR常用数据
官网👉DataResource
3. SMR分析基础
SMR(Summary-data-based Mendelian
Randomization,SMR)分析基于SMR软件完成,SMR分析使用smr-format格式文件,检验甲基化、基因表达和蛋白表达数量性状位点(mQTLs、eQTLs和pQTLs)与疾病或者性状之间关系的探究,用于探究疾病的发病机制。而HEIDI检验主要用于检验,基因snp介导的表型是否是由于连锁不平衡反应导致的。
4. SMR文件格式
SMR分析使用smr-format的格式,如eQTLGen来源的血液中基因表达的smr-format数据包含以下几个文件:
- cis-eQTLs-full_eQTLGen_AF_incl_nr_formatted_20191212.new.txt_besd-dense.besd
- cis-eQTLs-full_eQTLGen_AF_incl_nr_formatted_20191212.new.txt_besd-dense.epi
- cis-eQTLs-full_eQTLGen_AF_incl_nr_formatted_20191212.new.txt_besd-dense.esi
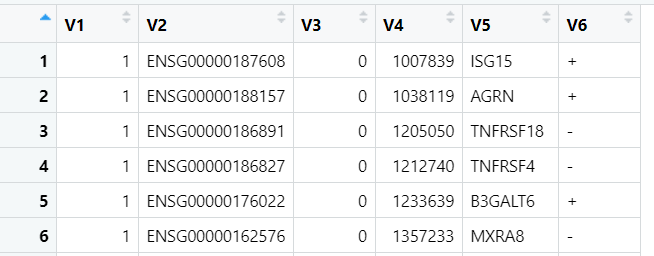
4.1 .epi文件
读取文件cis-eQTLs-full_eQTLGen_AF_incl_nr_formatted_20191212.new.txt_besd-dense.epi
如下是文件的前六行截图:
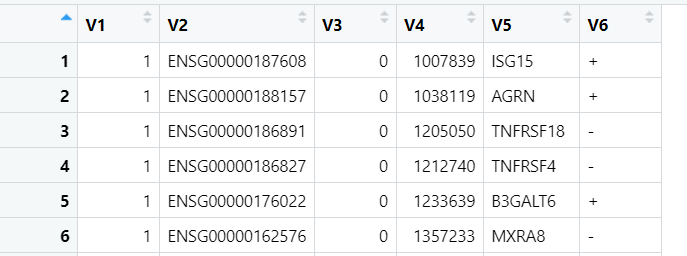
对文件中的每一列进行解读:
- V1: chromosome,对应探针的chromosome
- V2: probe ID,探针的ID (can be the ID of an exon or a transcript for RNA-seq data)
- V3: genetic distance (can be any arbitary value),可以直接设置为0,该列在SMR分析中不会被使用。
- V4: physical position,可以设置为基因起始碱基的中间值。
- V5: gene ID,可以是gene symbol或者gene ensembl id或者缺失值。
- V6: gene orientation,基因方向(此信息仅用于绘图)
4.2 .esi文件
读取文件cis-eQTLs-full_eQTLGen_AF_incl_nr_formatted_20191212.new.txt_besd-dense.esi
如下是文件的前六行截图:
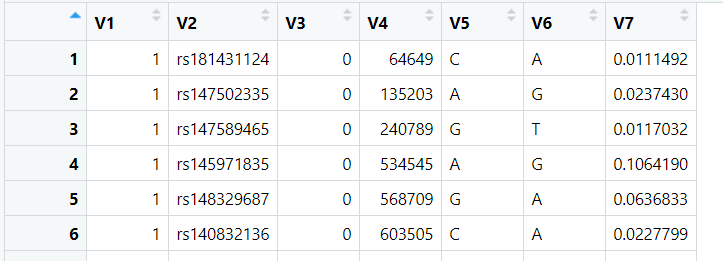
对文件中的每一列进行解读:
- V1: chromosome,SNP对应的chromosome
- V2: SNP,SNP的rsid
- V3: genetic distance (can be any arbitary value),可以直接设置为0,该列在SMR分析中不会被使用。
- V4: basepair position,SNP的碱基位置。
- V5: the effect (coded) allele,SNP的效应等位基因。
- V6: the other allele,SNP的参考等位基因。
- V7: frequency of the effect allele,SNP的效应等位基因频率
5. 探针ID列的重要性
在进行SMR分析的时候,尽可能用通用的代码去完成SMR分析,以减少工作量。 不管是DNA甲基化化,基因表达,还是蛋白表达,都可以理解为探针对应的QTLs数据与疾病GWAS数据的SMR分析。 所以我们第一步就是提出感兴趣甲基化区域、基因或者蛋白的探针ID,这对应在epi文件中的V2列。
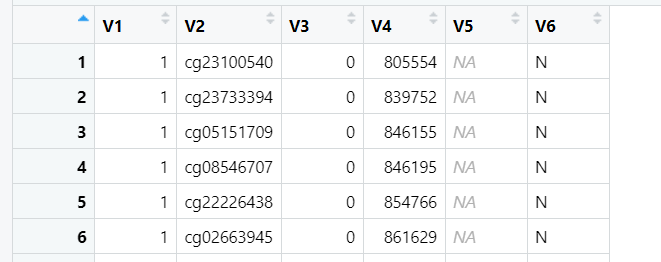
5.1 甲基化数据epi文件
如SMR官方提供的:McRae et al. mQTL summary data,探针列为cg开头的甲基化探针。DNA甲基化状态由Illumina HumanMethylation450芯片检测,所以这里的探针id为甲基化芯片中的探针ID。
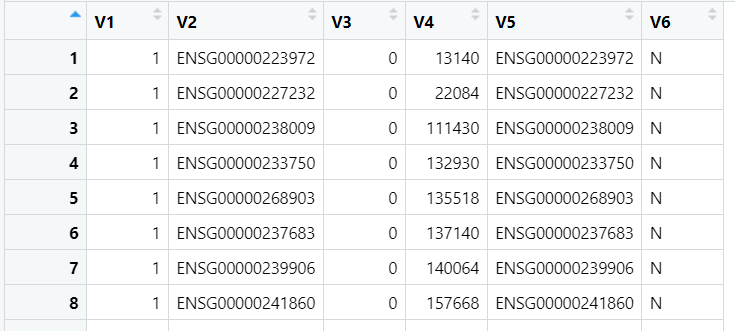
5.2 基因表达数据epi文件
如eqtgen提供的:cis-eqtls,探针V2列为基因的ensembl id,与v5列基因名是一一对应的关系。V5列可以是ensembl id(如SMR官方提供的Westra eQTL summary data),也可以是gene symbol(如GTEx提供的数据)。
5.3 蛋白表达epi文件
目前尚无官方提供的蛋白pQTLs文件,根据构建smr-format的格式,按照smr官方提供的构建besd方法1进行构建:1. Make a BESD file from eQTL summary data in ESD format
如下为将decode2021的蛋白数据构建为smr-format数据的epi文件:探针列为基因的ensembl id,v5列对应gene symbol。
6. 探针查询
6.1 基因或蛋白表达探针查询
通过前面的展示,可以知道,V2列探针列与V5列基因列是一一对应的关系,所以查询目标基因或者蛋白的表达对应的探针,可以通过如下两种方式:
- 直接通过基因的 Ensemble ID (如ENSG00000116288)
- 将基因或者蛋白名转换乘对应的 Ensemble ID(如将PARK7转换成ENSG00000116288)
6.2 甲基化探针查询
甲基化探针芯片有大概450k个探针,但是基因仅仅只有约2万个,所以甲基化探针与基因的关系是多对一的关系。 通过前面甲基数据epi文件,可知,V2列是探针名,形式为cgxxxxxx。V5列的基因名为空。查询目的基因或者蛋白对应的探针,可以通过如下方式:
- 根据目标基因的染色体区域,如基因的顺式调控区域,选择对应的甲基化探针。
6.3 查询探针代码演示
6.3.1 基于基因进行查询探针
以提取线粒体相关基因PARK7为例,展示基于基因提取对应的基因表达或蛋白表达探针
Ensembl_GRCh37[Ensembl_GRCh37$gene_name == "PARK7",]
# gene_id gene_name seqnames start end width strand source type gene_biotype
# 4594 ENSG00000116288 PARK7 1 8014351 8045565 31215 + ensembl_havana gene protein_coding
## 基于R包内置的数据集,完成匹配即可返回对应的Ensemble ID,即SMR分析中用到的探针信息V2列 6.3.2 基于基因的顺式调控区域查询探针
以提取线粒体相关基因PARK7为例,展示基于基因的顺式调控区域,提取对应的甲基化探针
# 以提取线粒体相关基因:PARK7为例
# 使用的函数是:smr_query_probes()
### 准备染色体区域信息
temp_gene <- "PARK7"
temp_anno <- Ensembl_GRCh37[Ensembl_GRCh37$gene_name == temp_gene,]
temp_chr <- gsub("chr","",temp_anno$seqnames,ignore.case = T)
temp_start <- temp_anno$start-1e6
temp_end <- temp_anno$end+1e6
query_chrompos <- paste0(temp_chr,":",temp_start,"-",temp_end)
# [1] "1:7014351-9045565"
### 进行目的基因的探针查询
smr_query_probes(smr_exe_path = "e:/smr-1.3.1-win-x86_64/smr-1.3.1-win.exe",
qtls_path = "z:/data/data-SMR/mQTLs/LBC_BSGS_meta_lite/mQTL_besd/LBC_BSGS_meta",
query_p = 5e-8,
query_chrompos =query_chrompos,
out_path = "./results/",
out_prefix = temp_gene)
# ✔ 查询数据输出到:./results/PARK7_chrompos_probes.txt
# $chrompos_path
# [1] "./results/PARK7_chrompos_probes.txt"